Spinalna mišićna atrofija
i Florian Tiefenböck, liječnik Ažurirano danaMaximilian Reindl studirao je kemiju i biokemiju na LMU u Münchenu, a član je uredničkog tima -a od prosinca 2020. godine. Upoznat će se s vama o temama medicinske, znanstvene i zdravstvene politike kako bi bile razumljive i razumljive.
Još postova od Maximilian ReindlFlorian Tiefenböck studirao je ljudsku medicinu na LMU München. Pridružio seu kao student u ožujku 2014. i od tada podržava urednički tim medicinskim člancima. Nakon što je dobio liječničku dozvolu i praktični rad iz interne medicine u Sveučilišnoj bolnici Augsburg, od prosinca 2019. stalni je član tima i, između ostalog, osigurava medicinsku kvalitetu alata
Još postova od Floriana Tiefenböcka Sav sadržaja provjeravaju medicinski novinari.
Spinalna mišićna atrofija ili skraćeno SMA rijetka je bolest u kojoj određene živčane stanice u leđnoj moždini umiru. Poticaji i impulsi iz mozga tada više ne stižu do odredišta: mišića. To uzrokuje gubitak mišića i paralizu. Postoje različiti oblici SMA. Najteži počinje u djetinjstvu. Novi tretmani obećavaju trajno poboljšanje zdravlja. Više o spinalnim mišićnim atrofijama pročitajte ovdje.
ICD kodovi za ovu bolest: ICD kodovi su međunarodno priznati kodovi za medicinske dijagnoze. Mogu se naći, na primjer, u liječničkim dopisima ili na potvrdama o nesposobnosti za rad. G12

Kratak pregled
- Što je spinalna mišićna atrofija? Skupina bolesti mišićne slabosti. Temelje se na smrti određenih živčanih stanica u leđnoj moždini koje kontroliraju mišiće (motorne neurone). Stoga su SMA među bolestima motoričkih neurona.
- Koji oblici postoje? Nasljedne spinalne mišićne atrofije uglavnom su SMA s određenim genetskim defektom na kromosomu 5 (5q-povezana SMA). Liječnici razlikuju četiri različita oblika: SMA tip 1 - tip 4. Osim toga, postoje sporadični oblici čija nasljednost nije sigurna.
- Učestalost: Rijetke bolesti; nasljedna SMA pogađa oko jedno novorođenče u 7000.
- Simptomi: trzanje mišića, progresivna slabost mišića, gubitak mišića, paraliza. Gradijenti se razlikuju ovisno o obliku SMA.
- Uzroci: Nasljedna spinalna mišićna atrofija tipa 1-4 posljedica je genetskog defekta na kromosomu 5, točnije na genu SMN1. Zbog toga tijelu nedostaje poseban protein, protein SMN. Taj nedostatak oštećuje motorne neurone u leđnoj moždini.
- Dijagnoza: Genetski pregled za izmjenjenu genetsku strukturu SMN -a, fizikalni pregledi, elektroneurografija, elektromiografija, testovi krvi (npr. CK)
- Liječenje: Moguća je genska nadomjesna terapija ili primjena lijekova modulatora spajanja. Popratna fizioterapija, logopedija, terapija boli i psihoterapija. Ako je potrebno, operacija na kralježnici. Plan liječenja ovisno o vrsti SMA.
- Prognoza: U slučaju nasljednog proksimalnog SMA, nove mogućnosti terapije imaju uzročni učinak i mogu imati pozitivan učinak na tijek bolesti. Rani početak liječenja ključan je. Liječenje još nije dostupno svakom pacijentu. Ako se ne liječi, djeca sa SMA tip 1 obično umiru unutar prve dvije godine. Očekivano trajanje života u tipovima 3 i 4 teško se ili ne smanjuje.
Što je spinalna mišićna atrofija?
U spinalnoj mišićnoj atrofiji (SMA), određene živčane stanice u leđnoj moždini umiru. Obično kontroliraju mišiće, pa stručnjaci te živčane stanice nazivaju motornim neuronima. Sukladno tome, SMA spadaju u takozvane bolesti motornih neurona.
U slučaju spinalnih mišićnih atrofija, zahvaćeni su donji (drugi) motorni neuroni, koji su svojim dodacima izravno povezani s mišićima. Kao rezultat oštećenja, manje ili više živčanih signala dopire do mišića. Mišići postaju sve slabiji i manji (trošenje mišića / atrofija mišića).
Liječnici razlikuju različite oblike spinalne mišićne atrofije. Daleko najveća skupina su nasljedni SMA, u kojima su zahvaćeni mišići blizu trupa (proksimalni). Temelje se na specifičnom genetskom defektu. Otprilike jedno od 7000 novorođenčadi razvit će ga.
Spinalna mišićna atrofija općenito je rijetka bolest. Ipak, to je druga najčešća autosomno recesivna nasljedna bolest, ali i najčešći uzrok smrti beba ili male djece zbog genetskog defekta.
Koje vrste spinalne mišićne atrofije postoje?
Liječnici razlikuju nasljedne (nasljedne) oblike SMA od sporadičnih. Druga klasifikacija spinalne mišićne atrofije odnosi se prvenstveno na mišićne skupine koje su prvo zahvaćene. Tamo su
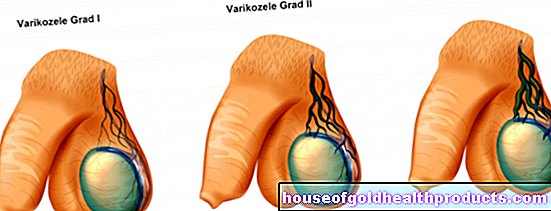
- Proksimalni SMA: S oko 90 posto oni čine najveću SMA skupinu. Simptomi počinju u mišićima blizu trupa, tj. Proksimalno.
- Neproksimalna SMA: Ovdje su najprije zahvaćene udaljenije mišićne skupine, poput šaka i stopala (distalna SMA). U daljnjem tijeku, ti se SMA mogu proširiti i na mišiće blizu sredine tijela.
- Posebni oblici (npr. Spinobulbarna mišićna atrofija tipa Kennedy)
Proksimalna spinalna mišićna atrofija
Nasljedne proksimalne spinalne mišićne atrofije uglavnom su bolesti koje se temelje na specifičnom genetskom defektu (SMA povezan s 5q, defekt na kromosomu 5). Oni su pak podijeljeni u četiri različita oblika. Klasifikacija se temelji na vremenu u kojem se pojavljuju prvi simptomi i na tijeku bolesti.
Spinalna mišićna atrofija Tip 1: Ovo je najčešći i najozbiljniji oblik SMA. Također se naziva "Werdnig-Hoffmannova bolest" ili "akutna infantilna SMA". Bolest obično počinje u ranom djetinjstvu. Slabost mišića utječe na cijelo tijelo - liječnici također govore o "disketnom sindromu dojenčeta" (od engleskog floppy = mlitavo, dojenče = dojenče, dijete). Većina neliječene djece sa SMA tip 1 umire prije nego što napune dvije godine.
Spinalna mišićna atrofija tip 2: Ovaj oblik SMA poznat je i kao "srednja spinalna mišićna atrofija" ili "kronična infantilna SMA". Prvi simptomi obično se javljaju prije navršenih 18 mjeseci. Oboljeli imaju ponekad značajno smanjeni životni vijek.
Spinalna mišićna atrofija tip 3: Također je poznata i kao "juvenilna spinalna mišićna atrofija" ili "Kugelberg-Welanderova bolest". Ovaj SMA obično počinje nakon navršenih 18 mjeseci i prije rane odrasle dobi. Mišićna slabost je blaža nego kod tipa 1 ili 2. Oni koji su pogođeni imaju tek neznatno smanjeni životni vijek.
Spinalna mišićna atrofija tip 4: Slična je SMA tipu 3, ali se pojavljuje samo u odrasloj dobi (obično> 30 godina). Međutim, slabost mišića je manje izražena i napreduje sporije nego kod SMA tipa 3.
Prijelazi između različitih verzija su fluidni. U nekim slučajevima to otežava jasno razgraničenje. Neke genetske predispozicije također igraju važnu ulogu u težini dotične bolesti.
Druge spinalne mišićne atrofije
Osim ovih proksimalnih oblika, postoje i drugi oblici spinalne mišićne atrofije. To uključuje, na primjer, rjeđe, također nasljedne distalne spinalne mišićne atrofije. Kod njih simptomi tipično počinju u mišićnim skupinama koje su udaljenije od tijela.
Nasljeđivanje nije zajamčeno u slučaju sporadične SMA. Osim toga, ne može se utvrditi obiteljska akumulacija. U literaturi to uključuje:
- Hirayama tip (maloljetni distalni SMA, bolest oko 15 godina, zahvaća mišiće ruku, obično zastaje čak i bez terapije, pa se čak može i poboljšati)
- Vulpian-Bernhard tip (također sindrom "mlatilice" s početkom u ramenom obruču, obično od 40. godine)
- Duchenne-Aran tip (u početku zahvaćeni mišići šaka, šireći se prema trupu tijela, obično nakon 30. godine)
- Peronealni tip (sindrom "mlati nogu", prvo na mišićima potkoljenice)
- Progresivna bulbarna paraliza (poremećaji govora i gutanja, pogađaju oko 20 posto pacijenata s amiotrofičnom lateralnom sklerozom)
Neki sporadični oblici SMA (sindrom "mlati ruka -noga", progresivna bulbarna paraliza) ubrajaju se u varijante amiotrofične lateralne skleroze (ALS) u stručnim krugovima. Ovaj se članak bavi prvenstveno nasljednim proksimalnim spinalnim mišićnim atrofijama.
Spinobulbarna mišićna atrofija
Spinobulbarna ili bulbospinalna mišićna atrofija (Kennedyjev tip, Kennedyjev sindrom) je nasljedna bolest. Često počinje u mladosti do srednje odrasle dobi. Ovaj poseban oblik SMA nasljeđuje se na X-vezan recesivan način i stoga utječe samo na muškarce (budući da muškarci imaju samo jedan X kromosom, u žena prevladava drugi, zdravi X kromosom koji bi nadoknadio nedostatak).
Česta je slabost mišića u mišićima blizu tijela na nogama i rukama ili ramenima, kao i u mišićima jezika i grla. Kao rezultat toga, pogođeni imaju probleme s govorom i gutanjem, na primjer. Također se žale na podrhtavanje, grčeve u mišićima i trzanje. Oboljeli muškarci također često imaju zakržljene testise i sterilni su. Osim toga, povećavaju se mliječne žlijezde (ginekomastija).
Spinobulbarna mišićna atrofija obično je spora. Očekivano trajanje života teško je ograničeno.
Kako prepoznati spinalnu mišićnu atrofiju?
Tipično za spinalnu mišićnu atrofiju su progresivna slabost mišića do paralize (pareze) i trzanje mišića. Kao posljedica oštećenja živaca, mišići više ne primaju električne impulse zbog čega se s vremenom smanjuju (mišićna atrofija). Točni znakovi i pritužbe ovise o odgovarajućem obliku. U sljedećem odjeljku razmatraju se simptomi nasljednog proksimalnog SMA.
Simptomi dječje spinalne mišićne atrofije tipa 1
Kod SMA tipa 1 simptomi se pojavljuju u prvih šest mjeseci života. Javlja se opća slabost mišića - to jest ona koja utječe na cijelo tijelo. Osim toga, smanjuje se napetost između mišića. Liječnici govore o hipotoniji.
U novorođenčadi se ta slabost mišića u početku očituje u tipičnom držanju nogu, koje podsjeća na ležeću žabu (držanje žabljih nogu). Noge su savijene, koljena okrenuta prema van, a stopala pod kutom prema unutra. Čak ni samostalno podizanje ili držanje glave obično nije moguće.
U poodmakloj dobi djeca sa SMA tip 1 ne mogu samostalno sjediti ili hodati. Mnoga djeca također ne mogu govoriti jer mogu biti zahvaćeni i mišići jezika.
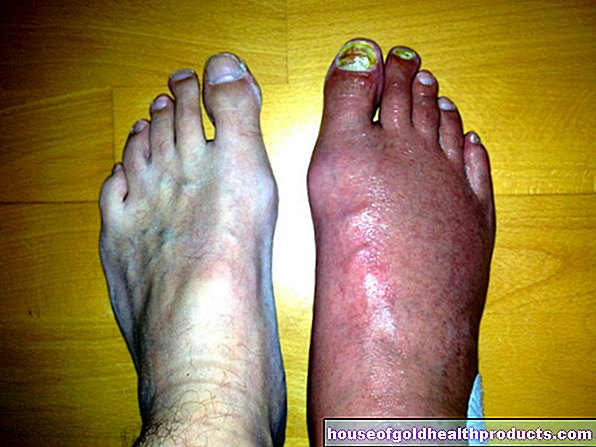
Druga karakteristika spinalne mišićne atrofije tipa 1 je oblik gornjeg dijela tijela: mišići prsa i leđa ne razvijaju se pravilno. Time gornji dio tijela dobiva oblik zvona (sanduk sa zvonom). Zbog slabog razvoja mišića u prsima i leđima, zahvaćeni zauzimaju pogrbljeno držanje.
Često postoji i sve veća zakrivljenost kralježnice (skolioza). Pogrbljeno naprijed i pogrbljeno držanje uzrokuje daljnje probleme s disanjem. Karakteristično je vrlo brzo i plitko disanje (tahipneja).
Simptomi srednje spinalne mišićne atrofije tipa 2
Spinalna mišićna atrofija tipa 2 obično uzrokuje samo simptome u dobi od sedam do 18 mjeseci. Oboljela djeca mogu sjediti sama, ali obično niti nauče stajati niti hodati. Mišićna slabost napreduje sporije nego kod tipa 1.
Kod SMA tipa 2 s vremenom se javljaju simptomi slični onima u teškom dječjem obliku, poput deformacije kralježnice. Zglobovi se ukrućuju zbog skraćenih mišića i tetiva (kontraktura). Drugi znakovi uključuju drhtavicu u rukama i trzanje mišića na jeziku.
Simptomi juvenilne spinalne mišićne atrofije tipa 3
Spinalna mišićna atrofija tipa 3 tipično se javlja nakon navršenih 18 mjeseci i prije 18. Oboljela djeca mogu samostalno sjediti, stajati i hodati. Međutim, slabost mišića, osobito u mišićima zdjelice i nogu, uzrokuje hodanje.
Tijekom nekoliko godina performanse se smanjuju: isprva oboljelima teško padaju sportske aktivnosti ili penjanje uz stepenice, ali na kraju je, također, teško nositi torbe za kupnju. Nakon mnogo godina, spinalna mišićna atrofija tipa 3 otežava ili čak onemogućava trčanje i bilo koji drugi napor, čak i kod starijih osoba.
Sveukupno, međutim, simptomi su manje izraženi nego kod dva druga oblika bolesti, tipa 1 i tipa 2. Za mnoge od oboljelih, kvaliteta života teško se narušava tijekom dužeg vremenskog razdoblja.
Simptomi spinalne mišićne atrofije odraslih tipa 4
Ovaj vrlo rijedak oblik progresivnog trošenja mišića počinje u odrasloj dobi, često od trećeg desetljeća života. U početku su zahvaćeni mišići nogu i kukova. Kako bolest napreduje, slabost mišića također se širi na ramena i ruke.
Klinička slika je slična manifestaciji juvenilnog SMA tipa 3. Međutim, progresivna mišićna slabost još je sporija nego kod SMA tipa 3.
Što uzrokuje spinalnu mišićnu atrofiju?
U spinalnoj mišićnoj atrofiji, drugi motorni neuroni u leđnoj moždini propadaju. To su živčane stanice koje svojim dodacima kontroliraju mišiće. Kao posljedica oštećenja ovih visoko specijaliziranih motornih neurona, manje električnih signala dopire do mišića nego što je to slučaj kod zdravih ljudi. Ako se mišićne stanice koriste manje i stoga manje stimuliraju, tijelo ih s vremenom razgrađuje.
Genetski defekt
U većini slučajeva, spinalna mišićna atrofija je nasljedna bolest (nasljedna SMA). Uzrok tipičnih proksimalnih SMA oblika su netočne informacije u genetskom sastavu pacijenta. Takozvani gen SMN1 na kromosomu 5 nije funkcionalan.
Gen SMN1 nosi informaciju - tj. Nacrt - za vitalnu molekulu proteina zvanu SMN. SMN je kratica za "Survival (of) Motor Neuron". Bez molekule proteina SMN, motorni neuroni s vremenom nestaju.
Istina je da u tijelu postoji i srodni gen SMN2, koji u načelu može "kompenzirati" nefunkcionalne genetske informacije SMN1. No to se obično događa samo u maloj mjeri. To znači da se gubitak funkcije gena SMN1 (ako se ne liječi) obično ne može u potpunosti nadoknaditi netaknutom kopijom gena SMN2.
Autosomno recesivno i autosomno dominantno nasljeđivanje
Genetski podaci neke osobe dostupni su u dva primjerka. Kao rezultat toga, svatko ima dvije kopije gena SMN1 - jednu od oca i jednu od majke. Proksimalne spinalne mišićne atrofije u djetinjstvu tipično se nasljeđuju kao autosomno recesivno svojstvo.
To znači da obje varijante gena (aleli) od roditelja moraju biti neispravne da bi se spinalna mišićna atrofija razvila u potomstvu. U slučaju recesivnog nasljeđivanja, roditelji nisu pogođeni jer, osim nefunkcionalnog, imaju i zdrav gen SMN1 koji kompenzira nedostatak.
Otprilike svaka 45. osoba vlasnik je ovog sustava za SMA. Par u kojem su oba partnera nositelji ima 25% rizika da dobije dijete s tom bolešću.
U nekoliko slučajeva u adolescenciji, spinalne mišićne atrofije osobito u odrasloj dobi također slijede autosomno dominantno nasljeđivanje. U slučaju dominantnog nasljedstva, defektni gen već se potvrđuje - i oboljeli postaju bolesni. Međutim, ovo nije prethodno spomenuti genetski defekt na kromosomu 5. Ovi 5q-povezani SMA-i uvijek se nasljeđuju na autosomno recesivan način.
Nasljeđivanje s drugim oblicima SMA
Neproksimalna spinalna mišićna atrofija također se može naslijediti. Posebni spinobulbarni oblik (Kennedyjev tip) nasljeđuje se recesivno putem spolnog kromosoma, X kromosoma (to utječe na varijante gena koje sadrže nacrt za spajanje muških spolnih hormona). U slučaju sporadičnih oblika, međutim, nasljeđivanje nije zajamčeno. Ovdje se malo zna zašto upravo drugi motorni neuroni nestaju.
Pregledi i dijagnoza
Dijagnozu spinalne mišićne atrofije obično postavljaju pedijatri, pedijatri specijalizirani za živčane bolesti (neuropedijatri) i specijalisti za bolesti živčanog sustava (neurolozi). Za preciznija pojašnjenja potrebna su različita ispitivanja. U slučaju SMA, genetski testovi i pregledi živaca i mišića posebno su važni.
Zbirka povijesti bolesti (anamneza)
Uz svaku bolest, liječnik prvo pita o simptomima koji su se javili i kako je do sada napredovao. Kod beba i male djece roditelji prijavljuju promjene i abnormalnosti u ponašanju djeteta. Posebno u slučaju nasljednih bolesti, liječnici se također usredotočuju na obiteljsku povijest bolesti.
Tjelesni pregledi
U osnovi, liječnik fizičkim pregledom djeteta utvrđuje abnormalnosti u motoričkom razvoju. Na primjer, provjerava mogu li djeca samostalno držati uspravno glavu, sjediti ili samostalno micati rukama ili nogama (ovisno o dobi).
Dopunski testovi vježbi provode se kod starije djece i odraslih sa sumnjom na spinalnu mišićnu atrofiju. Liječnik provjerava koliko dotična osoba može skupiti i koliko dugo može izdržati. Ispituje i izdržljivost.
Osim toga, liječnik testira reflekse koji su tipično oslabljeni ili ugašeni, osobito u slučaju izraženih spinalnih mišićnih atrofija. Da bi to učinio, čekićem lupka po različitim tetivama, na primjer po peti ili ispod koljena, te provjerava reakciju.
Genetske studije
Najpouzdanija metoda otkrivanja (nasljedne) spinalne mišićne atrofije je genetska analiza. Liječnici traže dokaze o promijenjenom (mutiranom) genu SMN1 i broju postojećih kopija SMN2.
Opće je pravilo dijagnosticiranje i liječenje (nasljednog) SMA -a što je prije moguće. Ovisno o obliku i dostupnom liječenju, na motorički razvoj može se pozitivno utjecati prije nego što se motorni neuroni leđne moždine nepovratno oštete.
Daljnja istraživanja na SMA -i
Ako se sumnja na SMA, liječnici često mjere brzinu provođenja živaca (elektroneurografija) i mišićnu aktivnost (elektromiografija). Ako je potrebno, pregledavaju i mišiće pomoću ultrazvuka (miosonografija) ili magnetske rezonancije (MRI).
Osim toga, liječnici dogovaraju krvne pretrage. Ako postoji spinalna mišićna atrofija, neki se parametri mogu promijeniti: na primjer, povećava se razina kreatin kinaze (CK, tipični mišićni enzim).
Liječenje spinalnih mišićnih atrofija
Liječenje spinalne mišićne atrofije složeno je. Dugo vremena uzročna terapija nije bila moguća za bilo koji oblik SMA. Međutim, napredak u medicinskim istraživanjima pruža liječnicima nove mogućnosti liječenja za temeljnu pomoć oboljelima od proksimalne SMA (defekt gena SMN na kromosomu 5).
Osim toga, liječnici se koncentriraju na ublažavanje simptoma i pružanje najbolje moguće podrške oboljelima (npr. Fizioterapija, respiratorna terapija, psihoterapija, eventualno operacija).
Medicinska terapija
Novi pristupi liječenju pacijenata kod kojih se SMA temelji na poznatom genskom defektu SMN interveniraju izravno u sam genetski materijal ili u daljnjoj obradi genetskih informacija.
Cilj je omogućiti tijelu pacijenta da samostalno proizvodi dovoljne količine proteina SMN, koji je ključan za motorne neurone.
Za spinalnu mišićnu atrofiju dostupne su sljedeće mogućnosti liječenja:
- Modulatori spajanja (Nusinersen, Risdiplam): Ovi lijekovi izravno interveniraju u daljnjoj obradi molekula RNA glasnika. Jačaju one procese koji isporučuju veću količinu SMN proteina iz netaknutog gena SMN2.
- Zamjenska terapija genom (Onasemnogene Abeparvovec): Ova terapija intervenira izravno u ljudski genom. Neispravna kopija gena SMN1 zamijenjena je u zahvaćenim stanicama vanjskim opskrbljenim, funkcionalnim genskim konstruktom.
Modulatori spajanja
U slučaju defekta gena SMN1, tijelo može alternativno proizvesti protein SMN iz srodnog gena SMN2. Zamjenski gen SMN2 "uskače", ali to nije dovoljno. Razlog: Proteini u SMN2 obično su prekratki i brzo se razgrađuju.
To je posljedica obrade odgovarajuće SMN2 glasničke RNA (SMN2 mRNA). On prenosi informacije o izgradnji iz genoma (DNA) na mjesta proizvodnje proteina (ribosome).
Da bi se to učinilo, prvo se čita gen SMN2 u genomu. Proizvodi se preliminarna SMN2 glasnička RNA. Između ostalog, mora se dodatno obraditi takozvanim spajanjem. Tek tada nastaje RNA zrelog glasnika. Posebni stanični kompleksi, ribosomi, zatim očitavaju zrelu glasničku RNA i tako proizvode protein SMN2. A upravo je to ono što je skraćeno i nestabilno, brzo se demontira i ne može preuzeti funkciju SMN1.
Da bi se to promijenilo, aktivni sastojci Nusinersen i Risdiplam utječu na daljnju obradu RNK preliminarnog glasnika. Kao rezultat toga, ti takozvani modulatori spajanja u konačnici povećavaju količinu upotrebljivih SMN proteina - i na taj način mogu osigurati odgovarajuću opskrbu.
Nusinersen
Lijek Nusinersen je takozvani "antisense oligonukleotid" (ASO). Odobrila ga je Europska agencija za lijekove 2017. godine. ASO su umjetno proizvedene i posebno prilagođene molekule RNK. Vežu se specifično i precizno na SMN2 glasničku RNK. Time se sprječava njihova daljnja pogrešna obrada u ljudskoj stanici.
Konkretno: Nusinersen sprječava pogrešno izrezivanje važnih informacija (egzon 7) iz RNK glasnika SMN2. Gdje se nalazi egzon 7 uzrokuje da tijelo naknadno proizvodi funkcionalniji SMN protein.
Nusinersen se daje putem lumbalne punkcije. To znači da se lijek ubrizgava u spinalni kanal štrcaljkom. Ova terapija se ponavlja u redovitim razmacima od nekoliko mjeseci. U prvoj godini liječenja oboljeli primaju šest, zatim tri doze godišnje.
Pacijenti obično dobro podnose lijek. Nusinersen dovodi do povoljnijih tijekova bolesti. Studije su pokazale da se mobilnost poboljšava kod mnogih pacijenata: u mnogim slučajevima bilo je moguće slobodno sjediti i samostalno okretati tijelo. Nuspojave i komplikacije temelje se, između ostalog, na lumbalnoj punkciji (npr. Glavobolja, infekcije moždanih ovojnica).
Risdiplam
Europska komisija odobrila je Risdiplam kao treći lijek protiv SMA povezane s 5q (tipovi 1-3 ili jedna do četiri kopije gena SMN2) u ožujku 2021. godine. Risdiplam se uzima svakodnevno kao otopljeni prah. Točna doza izračunava se na temelju dobi i tjelesne težine.
Za razliku od Nusinersena, Risdiplam nije "antisense oligonukleotid", već mala molekula. Ova molekula veže se na glasničku RNK za proteine SMN2 i stabilizira ih na ovaj način. Kao rezultat toga, stvaraju se funkcionalniji SMN proteini.
Uobičajene nuspojave Risdiplama uključuju gastrointestinalne tegobe, osip, groznicu i infekcije mokraćnog sustava.
Zamjenska terapija genima
Drugi pristup liječenju proksimalne spinalne mišićne atrofije oslanja se na ono što je poznato kao genska nadomjesna terapija. Neispravan gen SMN1 - polazište progresivne SMA - "zamjenjuje se" novom funkcionalnom kopijom gena.
Aktivni sastojak Onasemnogene Abeparvovec (AVXS-101), koji radi na ovom principu, dobio je uvjetno odobrenje za stavljanje u promet male djece i djece od Europske agencije za lijekove (EMA) u svibnju 2020. godine.
Prema podacima EMA -e, lijek se može koristiti za SMA tip 1. U svim drugim oblicima SMA bolesti, genetske karakteristike (broj kopija SMN2) odlučuju je li zamjenska terapija genom opcija.
Uz Onasemnogene Abeparvovec, funkcionalna kopija humanog gena SMN1 uvodi se u zahvaćene stanice leđne moždine i moždanog debla. To čine određeni virusi koji služe kao "trajekti" za novi genetski materijal-takozvani adeno-povezani virusni vektori (AAV vektori).
Konstrukti vektorskih gena daju se jednom kao infuzija kroz venu u krvotok i odatle se distribuiraju po cijelom tijelu. Zbog još nedovoljno razvijene krvno-moždane barijere u male djece, ti vektori mogu ući i u tkivo leđne moždine.
Preferirano vežući ove vektore za posebne površinske strukture motornih neurona, oni prvenstveno preuzimaju genetski materijal kako bi zatim neovisno proizveli protein SMN.
Liječenje može poboljšati motoričke funkcije i dovesti do trajnog razvojnog uspjeha (npr. Sjedenje, puzanje i hodanje bez podrške).Tijekom liječenja vrijednosti jetre ponekad se mogu značajno povećati, ali se broj krvnih pločica može smanjiti. Groznica i povraćanje su također česti. Kako bi se smanjile nuspojave, pacijentima se daju kortikosteroidi ("kortizon") nekoliko tjedana.
Motorni razvoj primjeren dobi općenito je moguć samo ako je genska terapija započela presimptomatski. Liječenje se odvija u specijaliziranim centrima za neuromišićno liječenje.
fizikalna terapija
Fizioterapija je i dalje važan stup liječenja SMA -e. Ne može se svaki oblik SMA liječiti novim pristupima liječenju. Redovita terapija vježbama osmišljena je za održavanje tjelesnih sposobnosti i usporavanje razgradnje mišića.
Fizioterapeut pasivno pomiče dijelove tijela koji su već paralizirani. Aktivne sekvence pokreta, pak, treniraju se kako bi podržale pokretljivost i snagu mišića. Masaža ili tretmani toplinom i hladnoćom također mogu pomoći. Oni također služe za opuštanje i pod određenim okolnostima usporavaju daljnju degeneraciju.
Govorna terapija
U nekim slučajevima SMA utječe na mišiće govora i gutanja. Tada pomaže logopedska vježba. Potiče djecu da nauče govoriti. Čak i kod starijih pacijenata to obično može usporiti pogoršanje govora. Logopedi također treniraju pravilno gutanje.
I fizioterapeuti i logopedi podržavaju oboljele ciljanom terapijom disanja.
Liječenje protiv bolova
Terapija boli igra važnu ulogu, osobito u naprednijim stadijima bolesti. Liječnici koriste lijekove protiv bolova kako bi smanjili patnju oboljelih.
kirurgija
Budući da spinalna mišićna atrofija može dovesti do ozbiljnog zakrivljenosti kralježnice (skolioza), liječnici ponekad razmišljaju o operaciji. Pritom posebno ukrućuju kralježnicu.
Time se zahvaćenima daje (određena) dodatna stabilnost trupa, koja ne samo da omogućuje uspravnije držanje, već štiti i kosti i zglobove. Operacija kralježnice također može pomoći protiv progresivnih problema s disanjem.
Psihoterapeutska njega
Neuromuskularne bolesti poput spinalne mišićne atrofije predstavljaju veliki psihološki stres.Pacijenti i rodbina dijagnosticiraju dijagnozu na individualnim i grupnim sastancima vođenim psihoterapijom te razvijaju strategije za bolje suočavanje s bolešću.
Grupe za samopomoć i predstavnici pacijenata također nude važnu podršku. Oni informiraju, savjetuju i podržavaju oboljele i njihovu rodbinu kako bi se nosili sa izazovima SMA bolesti.
Mogućnosti oporavka od spinalnih mišićnih atrofija
Ako postoji spinalna mišićna atrofija, prognoza prvenstveno ovisi o odgovarajućem obliku. Što se simptomi kasnije pojave, tijek će biti bolji. Osim toga, što ranije liječnici dijagnosticiraju spinalnu mišićnu atrofiju, prije mogu započeti odgovarajuće mjere liječenja, čak i prije nego što su motorni neuroni nepovratno oštećeni.
Nove mogućnosti liječenja kroz modulatore spajanja i nadomjesnu terapiju genima imaju veliki potencijal u liječenju proksimalnog SMA - osobito na početku (vrlo) ranog liječenja. Međutim, još uvijek se čekaju podaci za pouzdanu dugoročnu prognozu. Samo daljnje studije i usko povezana zapažanja o sigurnosti lijekova mogu pružiti daljnju sigurnost tijekom sljedećih (mjeseci i) godina. S novijim lijekovima barem se može zamisliti dugotrajna kontrola bolesti ili čak izlječenje.
SMA tip 1 općenito je ozbiljna bolest. Djeca koja razviju SMA tip 1 imaju (neliječeno) vrlo ograničen životni vijek. Brzo rastuća slabost mišića po cijelom tijelu također utječe na disanje. Posljedice su akutna upala pluća, pa čak i zatajenje disanja. Oboljela djeca umiru u prvih nekoliko godina života.
Prognoza je nešto bolja za SMA tip 2. Očekivano trajanje života varira ovisno o točnoj ozbiljnosti bolesti: neki umiru u djetinjstvu, ali većina njih doseže mlađu odraslu dob. Prije ili kasnije - ako se želi - disanje mora biti podržano u težim oblicima. Oboljeli ostaju mobilni uz pomoć invalidskih kolica.
Kod SMA tipa 3 prognoza je znatno bolja - osobito ako se prvi simptomi pojave kasno. Učinak se postupno pogoršava tijekom nekoliko godina. U starijoj dobi možda će biti potrebna invalidska kolica ili čak stalna njega. Očekivano trajanje života teško je ograničeno spinalnom mišićnom atrofijom tipa 3.
Spinalna mišićna atrofija odraslih (tip 4) čak je sporija od tipa 3. Ljudi obično imaju normalan očekivani životni vijek.